InquilineKea
Posts 2
Comments53
Was Cremieux considered to be one of the HBD/eugenics people?
[Twitter also kept pushing him into my feed even though I didn't follow him...]
If you look at the schedule of events(https://schedule.manifest.is/Manifest?view=text), Jonathan Anomaly is the only person who is very clearly HBD (the Collins allegedly, and then there's Stephen Hsu, who's HBD-adjacent). From the surface, it doesn't look like the HBD participants got any extra-special visibility? (though they just might be especially conspicuous in how unfiltered they are in bringing the topics out ) Gene Smith is there and has a post on gene editing for intelligence but AFAIK has never been associated with the HBD crowd (and doesn't get into the controversial social aspects) [also, gene-editing, unlike eugenics, can help empower the "have nots"].
Brain organoids are a way to quickly get functional/morphological significance readouts of intelligence-related genes (or genes related to other functions), so they are useful as a way of studying intelligence.
https://www.frontiersin.org/journals/science/articles/10.3389/fsci.2023.1148127/full
As a huge, huge moonshot, one could investigate avian brain organoids as an alternative substrate for intelligence (they are way more space-efficient than mammalian brains, and potentially could do way more compute in a small (manageable) volume if appropriately cultured and unbounded...
As N=1, I became vegetarian in 7th grade. Extreme levels of disgust-sensitivity [PETA is good at this] fueled this decision and I feel like I want to puke if I taste meat (I suspect disgust-sensitivity isn't as big of a factor in utilitarian EAs who go veg*)...
The factors were:
- personal interactions with chickens I had when staying over at a Woodinville farm (and then personal interactions with a Bellevue Chinese restaurant where I felt angered at how insensitive people were killing the aquarium fish right on site just for dishes (I was seeing murder at site). I stopped eating fish/chicken first, before I stopped eating all the other meats.
- PETA [and other veg*n advocacy sites] successfully grossing me out (eg "poop-contaminated meat") to the extent that I could not stand the taste of meat anymore. I did try fish once for the hell of it last year (when food was low) and still disliked the taste
- I was kind of obsessed with animals (Wildlife Factfile, Grolier, watching Wild Discovery, a book on fish my mom once checked out of the Redmond library which gave me intense appreciation of all the different species of fish [1]) and environmental protection even in my very early years [eg I was a super-notorious environmentalist in 2nd grade, and started freaking out about climate change in 5th grade due to an unusually hot summer in Denver]. This is partly because of the green (or at least pro-animals) messages that are sent in a lot of children's reading material
- Intense rage/hypocrisy at how humans were treating animals in general. Also me being mad at my parents for how little they cared [even though I didn't mind it as much in friends if they didn't go veg]... (contrarianism against parents...)
- a Seattle Times article about calorie restriction (and vegetarian diets are very obviously lower in calories). Along with all the health reasons (eg no saturated fat). Health factors were not the primary factors back then, but I would say that they are overwhelmingly the reason I now stay vegetarian (ofc to me there are still a million arguments for and almost none against, so it doesn't make sense for me not to be veg*).
During 7th grade, I remember all the vicious vegetarianism debates on Age of Kings Heaven/HeavenGames [lol @ f18fett's weird arguments against vegetarianism, as well as all the right-wingers who were against it], especially when I joined in with the other compassionate/left-wing forumers on those massive debate threads.
Going vegetarian + going to college early were the two kinds of contrarianism that I took that I am proudest of.
[1] Never realizing that it would cause intense amounts of family fighting when I finally took the plunge and became vegetarian. Ofc, this was not the end of family fighting over food (when I later also decided to turn against rice)
(a good number of people feel just as fine on a vegan diet as a non-vegan one [example] - most of the potential "costs" are in terms of energy/vitality which they will feel if they feel it). I am ovo-vegetarian and I have reasonably high iron levels myself. From the limited studies available, vegans have lower ACM than even vegetarians do.
It's important for people to get an ION Panel to test for amino acid deficiencies (these are hard to order w/o a doctor)
Scientists found three genes linked with vegetarianism and another 31 genes that are potentially associated.
Several of these genes, according to the study, are involved in lipid (fat) metabolism and/or brain function including two of the top three (NPC1 and RMC1).
“My speculation is there may be lipid component(s) present in meat that some people need. And maybe people whose genetics favor vegetarianism are able to synthesize these components endogenously,” Dr Yaseen said.
=====
4.2. Long Term Health Outcomes
Estimating the long-term health outcomes of eating certain things is difficult because food is highly bound up in the culture we live in and culture correlates to just about every health outcome you could possibly imagine. Even less conveniently, nutritional science is highly anti-inductive; if a particular food group is identified as being healthy people with an interest in being healthy flock to that food group, and people with an interest in being healthy are likely to be healthy for a bunch of reasons regardless of diet.
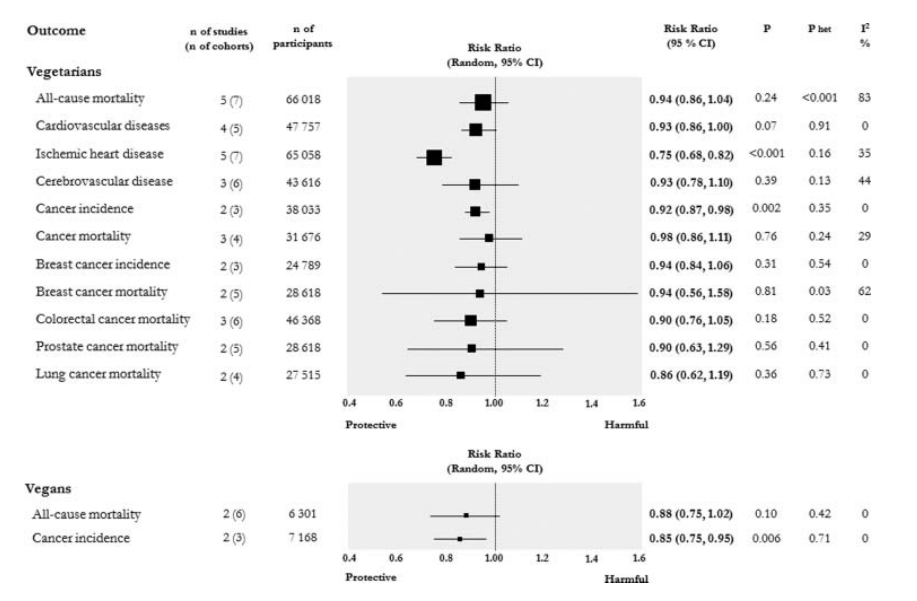
So here’s a nice headline result: vegetarians have less heart disease with extremely high certainty, and probably less cardiovascular disease and cancer too. Most of the studies in that meta-analysis have had some of the really obvious stuff adjusted away (race, income, etc.) but not all studies adjust for all confounders, and we should be cautious about trusting studies that ‘adjust for confounders’. If you ignore confounders then the answer is clear; eating vegetarian is good for you in every single way we can measure (including, possibly, circulating testosterone in defiance of stereotypes about meat eaters!).
If you are interested in confounders: There are a handful of cool natural experiments, taking groups with reasons to eat certain food but not bother with the associated healthy lifestyles, which are the closest we are likely to come to a true experiment in this area. In particular, the American Adventist Health Studies are pretty much state of the art in the field from what we can see. Adventists have quite unique dietary habits, brought about by religious prohibitions on certain foodstuffs which some Adventist churches follow and some don’t. Consequently, if you are an Adventist you are functionally ‘randomized’ into different food-eating conditions depending on which church you attend, and this randomization can be exploited by researchers.
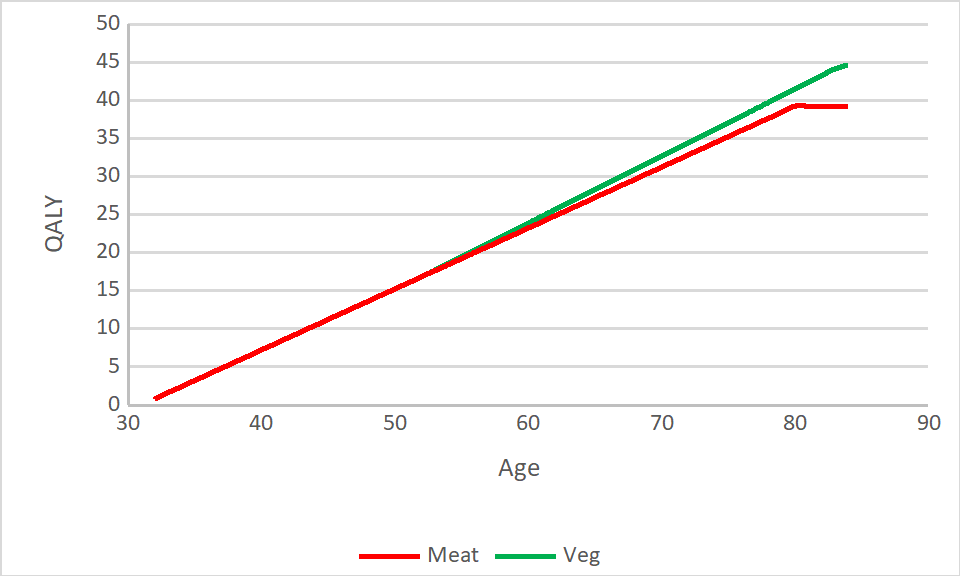
Based on the Adventist Health Studies, a vegetarian diet increases life expectancy by around 3.6 years. The less meat you eat, the healthier your BMI and the less likely you are to get diabetes.
Overall we might expect lacto-ovo vegetarians to have a health related quality of life around 10% better than a meat eater, with most of this benefit being apparent 20 years after making the switch to a vegetarian diet.
You could complicate this picture a lot (especially by introducing future discounting) but we think the general principle that if you value life-years towards the end of your life you should likely go vegetarian is well demonstrated by the data:
One final point on how meat might affect your lifespan; there is a growing awareness of the fact that industrially produced meat is an ideal breeding ground for zoonotic disease, and that those diseases can mutate and jump to humans very quickly. Previous pandemics such as H1N1 (‘swine flu’) and H5N1 (‘bird flu’) may have originated with farmed animals, and were rapidly spread by the close contact of unhealthy animals and global nature of the meat supply chain. At the margin, eating meat probably increases the probability of a global pandemic but there isn’t good evidence on how much your individual consumption affects things at the margin.
In the model we take the Adventist study result at almost face value, estimating that eating vegetarian will increase your lifespan by 3 years, and include constant low costs due to possible nutritional deficiency and moderate benefits to health that appear later in life.
There are many neurofeedback clinics and companies like Peak Brain Institute (they track brain waves and some people report drastic benefits, esp w/cognitive control, in response). The studies could be higher-quality and attract a broader range of people. They could also be way more widespread (also see what Neurable/Neurosity are doing). I tried neurofeedback once and can certify that I did feel very differently afterwards (in a good way!) but these protocol are usually very expensive (near-term AGI makes this matter way less). Many neurofeedback protocol try to increase the ratio of "high cognitive control" brainwaves (like gamma) over "low cognitive control" brainwaves (like the default mode network ones involves in daydreaming or that let mind wandering/outside stimuli overwhelm one's ability to have consistent plans that stay coherent through overly emotional stimuli)
Alex Milenkovic is studying the brainwaves of gamers! It's an important population b/c it is so easy to study+draw data out of.
I know Tomas Roy (at the Geneva Neurocenter) has special neurofeedback protocol for people with ADHD (one of his grad students investigates it) - and it is tested/scientific in a way that other protocol are not (though other protocol/montages record more data). I tried out one of them. Another professor at the institute, Daphné Bavelier, studies gamers.
I know some people who have set up a transcranial magnetic stimulation machine.
Jhourney is a startup (with super-legit people) focused on neurofeedback=> meditation.
On the topic of more general mind enhancement/cyborgism, see https://www.lesswrong.com/posts/vEtdjWuFrRwffWBiP/we-have-to-upgrade?commentId=7PBCydEDR8zptmLQY
I know many people have dissociated themselves from the nootropics community b/c none have been individually shown to on average be more effective than modafinil, but combinations can make a difference and there are some people who market them and make it frictionless for people to try/test them.
Reducing brain aging rate is also more tractable on average than increasing intelligence (esp b/c most people on Western diets have brains that shrink way faster than they should) => and is the most important way to maximize a brain's integrated lifetime total compute, and there are many ways to reduce this.
https://www.sciencedirect.com/science/article/pii/S2451902224003811#bib121
^enhancing equanimity through tFUS is very low-hanging fruit for improving QALYs and overall human lived experience (and is upstream of better decision making and all other good outcomes)
@NickCammarata is also v. into this, says that helping people there out >>> undergrad research just to get into neuro positions