Comments
What’s your theory for why the status quo tends to be wastewater?
What’s your theory for why the status quo tends to be wastewater?
For qPCR or other targeted detection approaches wastewater has quickly become a very common sample type, mostly because (a) it was very successful for covid, (b) a single sample covers hundreds of thousands of people, and (c) it's an 'environmental' sample so it's easy to get started (no IRB etc). And targeted detection is generally sensitive enough that the low concentrations are surmountable.
There isn't really a status quo for metagenomic monitoring: everything is currently in its early stages. There are academics collecting a range of samples and metagenomically sequencing them, but these don't feed into public health tracking, partly because they're not running their sequencing or analysis in a way that would give the low sample-to-results times you'd need from a real-time monitoring system.
Thanks to Michael Bryan, Mike McLaren, Simon Grimm, and many folks at the NAO for discussion that led to this tool and feedback on its implementation and UI.
The NAO works on identifying potential pandemics sooner, and most of our work so far has been on wastewater. In some ways wastewater is good—a single sample covers hundreds of thousands of people—but in other ways it’s not—sewage isn’t the ideal place to look for many kinds of viruses, especially respiratory ones.
We’ve been thinking a lot about other sample types, like nasal swabs, which have the opposite tradeoffs from wastewater: a single sample is just one person, but it’s a great place to look for respiratory viruses. But maybe then your constraint changes from whether you’re sequencing deeply enough to see the pathogen to whether you’re sampling enough people to include someone while they’re actively shedding the virus.
The interplay between these constraints was complicated enough that we decided to write a simulator, which then offered an opportunity to pull together a lot of other things we’ve been thinking about like the effect of delay, the short insert lengths you get with wastewater sequencing, cost estimates for different sequencing approaches, and the relative abundance of pathogens after controlling for incidence. We now have this in a form that seems worth sharing publicly: data.securebio.org/simulator.
The simulation is only as good as its inputs, and some of the inputs are pretty rough, but here’s an example. Let’s say we’re willing to spend ~$1M/y on a detection system looking for blatantly genetically engineered variants of any known human-infecting RNA virus. We’re considering two approaches:
We’d like to know how many people would be infected (“cumulative incidence”) before the system can raise the alarm.
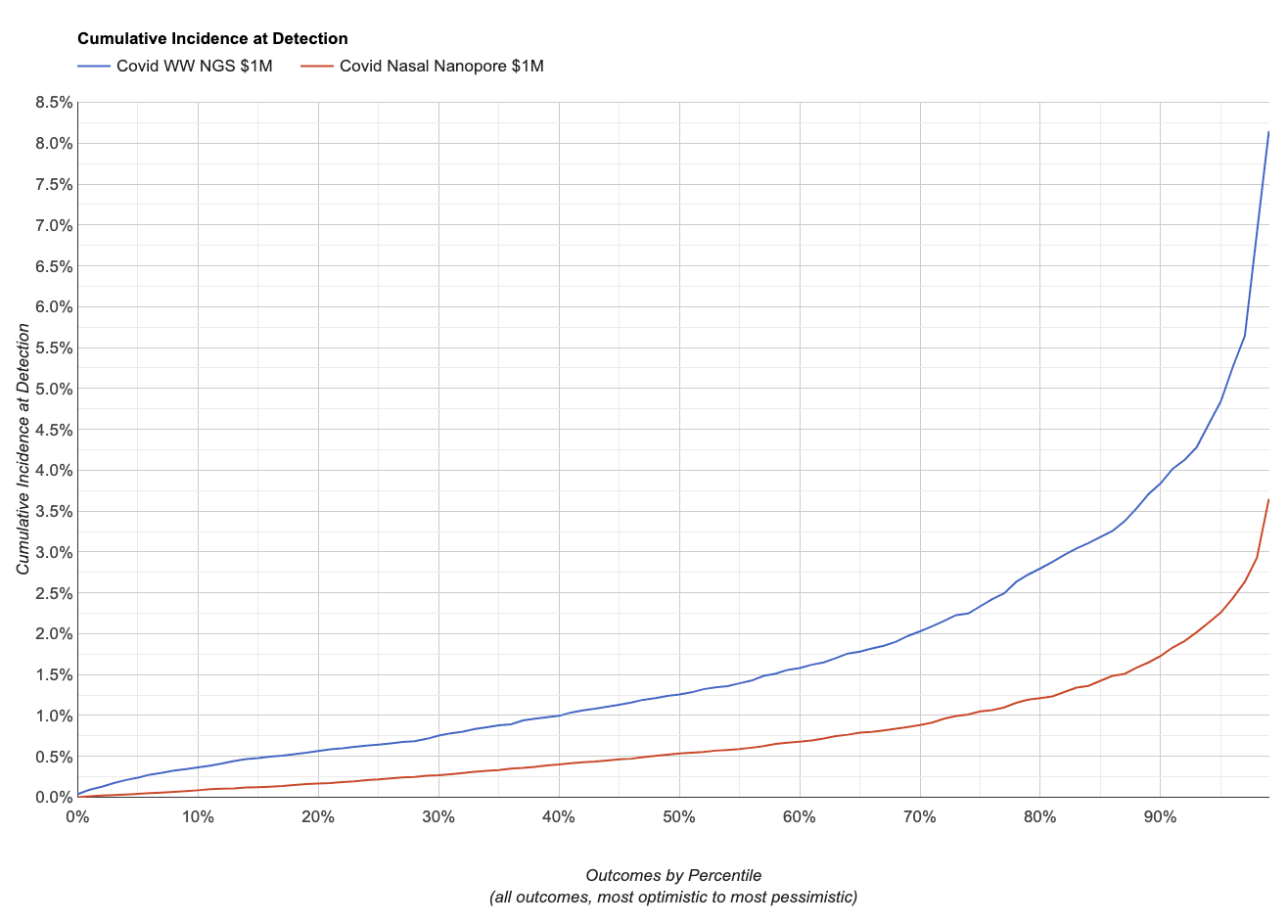
If the virus happens to shed somewhat like SARS-CoV-2, here’s what the simulator gives us:
A higher cumulative incidence at detection means more people have been infected, so on this chart lower is better. Given the inputs we used, the simulator projects Nanopore sequencing on nasal swabs would have about twice the sensitivity as Illumina sequencing on wastewater. It also projects that the difference is larger at the low percentiles: when sequencing swabs there’s a chance someone you swab early on has it, while with wastewater’s far larger per-sample population early cases will likely be lost through dilution. You can explore this scenario and see the specific parameter settings here.
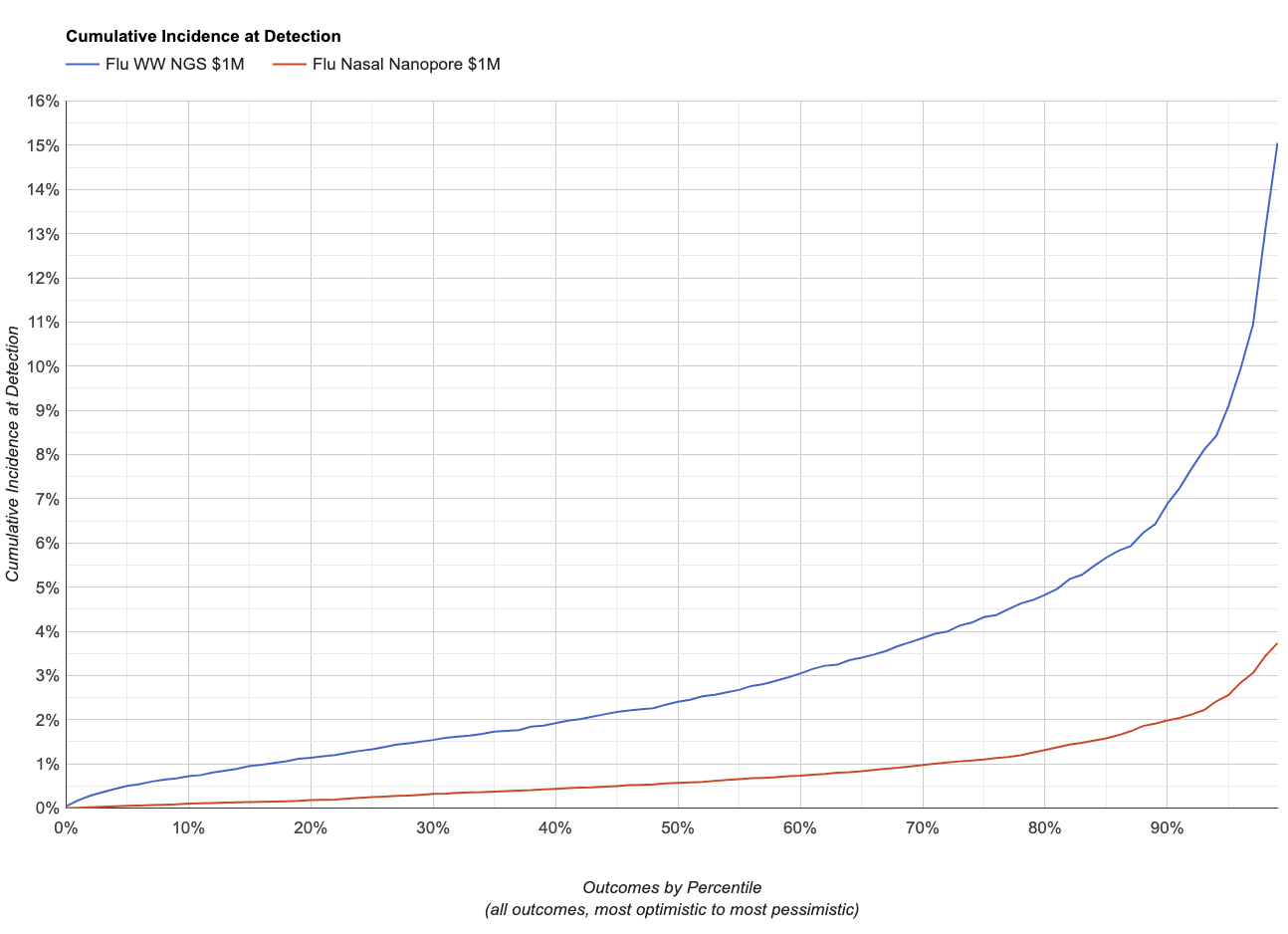
Instead, if it sheds like influenza, which we estimate is ~4x less abundant in wastewater for a given incidence, it gives:
This makes sense: if the pathogen sheds less in wastewater the system will be less sensitive for a given amount of sequencing.
On one hand, please don’t take the simulator results too seriously: there are a lot of inputs we only know to an order of magnitude, and there are major ways it could be wrong. On the other hand, however, Jeff adds that it does represent his current best guesses on all the parameters, and he does rely on the output heavily in prioritizing his work.1
If you see any weird results when playing with it, let us know! Whether they’re bugs or just non-intuitive outputs, that’s interesting either way.


Nice, good idea and well implemented!
In terms of wastewater being good for getting samples from lots of people at once and not needing ethics clearance, but being worse for respiratory pathogens, how feasible is airborne environmental DNA sampling? I have never looked into it, I just remember hearing someone give a talk about their work on this, I think related to this paper: https://www.sciencedirect.com/science/article/pii/S096098222101650X
I assume it is just hard to get the quantity of nucleic acids we would want from the air.
Flagging this for @Conrad K. - this seems like perhaps a better version of what you were considering building last year I think? If you have time you might have useful thoughts/suggestions.
I played around with the simulator a bit but didn't find anything too counterintuitive. I noticed various minor suboptimal things, depending on what you want to do with the simulator some of these may not be worth changing:
And the problem becomes worse if for whatever reason you run lots of scenarios, where the whole bottom half of the graph disappears:
Thanks for the feedback!
The values in the list aren't drawn from a parametrized distribution, they're the observed values in a small study.
Done!
Fixed!
This was due to me not testing on monitors that had that aspect ratio. Whoops! Fixed by allowing you to scroll that section.
We've done a fairly thorough investigation into air sampling as an alternative to wastewater at the NAO. We currently have a preprint on the topic here and a much more in-depth draft we hope to publish soon.
Oh nice, I hadn't seen that one, thanks!
Thanks for the tag @OscarD, this is awesome! I'd basically hoped to build this but then additionally convert incidence at detection to some measure of expected value based on the detection architecture (e.g. as economic gains or QALYs). Something way too ambitious for me at the time haha, but I am still thinking about this.
I definitely want to play with this in way more detail and look into how it's coded, will try and get back with hopefully helpful feedback here.