Correctly designed and with optimised protocols, LAMP is a cheaper, faster, more robust and thus better scalable alternative to PCR with comparable accuracy.
The molecular mechanism of LAMP is significantly harder to understand than PCR.
Why I think LAMP isn’t more widely used:
Designing and optimising a LAMP assay is much harder than conducting a pre-designed assay.
LAMP can outperform PCR specifically in diagnostics, but not in many other applications of PCR, and may thus be less well known.
PCR is particularly well-established, especially in the industrialised world, and there seems to be significant potential for catching up on establishment and standardisation of LAMP protocols through further research.
Offering cheaper testing methods than PCR, especially in low-income countries, might not pose enough commercial incentives for the market to fill the gap.
Pandemic preparedness is generally underfunded, and state actors are slow to implement new methods, indicating a need for public advocacy.
1. Introduction
Past pandemics have killed up to ~10% of the world population[1] and have severely affected human wellbeing, society and economies. Catastrophic pandemics could threaten the existence of present-day humanity and thus many future generations. The risk of catastrophic pandemics has recently increased with the advent of advanced artificial intelligence and thus a greater risk for engineered pathogens with enhanced pandemic potential. Preventing catastrophic pandemics is therefore one of the most pressing challenges of our time[2].
A key aspect of pandemic prevention is rapid and accurate detection of infections to contain local outbreaks and prevent them from growing into epidemics or (catastrophic) pandemics. The most accurate way of detecting infections to date is to amplify (i.e. to make copies of) pathogen-specific genetic material from patient samples and visualise the result to determine the presence or absence of the pathogen.
The first method described for amplifying genetic material is polymerase chain reaction (PCR) and was discovered by Kary B. Mullis in 1993[3]. To this date, PCR is the gold standard for DNA amplification and is used in biolabs around the world for research with small amounts of target DNA. Even though many modifications have been made to PCR to improve its accuracy and adjust it to more specialised purposes, the basic method is still the same as 30 years ago. While PCR is highly accurate, it is comparatively slow, requires specialised lab equipment and training and is thus expensive.
As pandemic preparedness and response are global concerns, there is a critical need for low-cost and low-tech, yet highly reliable and scalable testing methods, especially for low- and middle-income countries.
In 2000, Tsugunori Notomi and colleagues published another DNA amplification method called loop-mediated isothermal amplification (LAMP)[4]. It is faster, more robust, and cheaper than PCR and might thus be a better method to detect infections. However, having existed for almost 25 years, it has not developed into the new gold standard for DNA amplification and is still fairly unknown to the scientific community[5].
In this report, I give simple explainers for PCR and LAMP used for pathogen detection and work out their differences. I focus specifically on the key goals for biosecurity technology development laid out in “A framework for technical progress in biosecurity”, which are for methods to be fast, general, cheap, robust and scalable. I summarise current applications of LAMP, discuss why it is not more widely used and elaborate potential for adopting it more widely.
The report is aimed at readers who are interested in preventing (catastrophic) pandemics and who might have skills to further the development and wider adoption of LAMP. I expect readers with such skills to come from various backgrounds, which is why the report is set out to be understandable without a background in biology. Feel free to skip any parts that you are already familiar with.
2. PCR & LAMP for infectious disease diagnostics: How they work & key differences
This section describes the basic versions of PCR and LAMP used for infectious disease detection, and summarises the key differences.
First, the relevant basics on the structure and amplification of DNA that are relevant for understanding the following explanations:
DNA is a molecule that consists of two complementary strands that are held together by (weak) hydrogen bonds. The two strands can be separated by heating; this is called denaturation. Denaturation of DNA begins at 60-65°C.
There are four different nucleotide bases that form DNA: Adenine, Thymine, Guanine and Cytosine. In a complementary DNA double-strand Adenine pairs with Thymine and Guanine with Cytosine.
Each DNA single strand is made of nucleotides that are chained together via firm, covalent bonds that withstand DNA denaturation.
DNA polymerase is an enzyme that can add complementary nucleotides to a DNA single strand to synthesise a new, second strand and thus amplify DNA. DNA polymerases need a piece of double-stranded DNA (dsDNA) as a starting point (see “primer design” below). From this starting point they move along the DNA strand and add complementary nucleotides one by one.
Each DNA single strand has two different ends, the 3’- and the 5’-end (read: three-prime and five-prime). The DNA polymerase can only work in one direction, starting at a 3’-end of a primer.
Here’s what PCR and LAMP have in common:
Identification of a target DNA sequence which is unique to the pathogen of interest. The genetic sequence of the pathogen must be known, i.e. have been previously sequenced for this step.
Primer design | Primers are short DNA pieces (~20-40 nucleotides long) that are complementary to the DNA on both sides of the target sequence. After denaturing the DNA, a primer binds to its complementary sequence on a DNA single strand and provides a starting point for the polymerase. This step determines which part of DNA will be amplified, hence different primers are needed for different DNA targets.
Adding reagents | Besides target DNA, primers and polymerase, everyDNA amplification needs nucleotide building blocks and some further additives (buffer, certain ions the polymerase needs to work, etc.). Further additives can help improve amplification specificity.
DNA amplification reaction | The sample is incubated at the suitable temperature(s) providing the conditions for the DNA polymerase to do its work.
Visualisation of the result | The final step to determining whether a patient is infected with the pathogen of interest is to check whether the target DNA has been amplified in the previous reaction and was thus present in the initial patient sample.
PCR and LAMP differ most strongly in the amplification reaction itself, as well as the primer design and the result visualisation.
2.1 PCR
I will explain PCR first, as the principle is easier and helpful for understanding LAMP.
Primer design | One pair of primers (forward, backward) is needed for PCR.
Figure 1: Primers for PCR.
DNA extraction | PCR is quite sensitive against contaminants present in test samples (nasal/pharyngeal swab, blood, saliva etc.). Therefore, a DNA extraction step is required prior to the actual PCR reaction. There are various DNA extraction kits on the market, the fastest ones take ~20 minutes, slower ones can take a few hours.
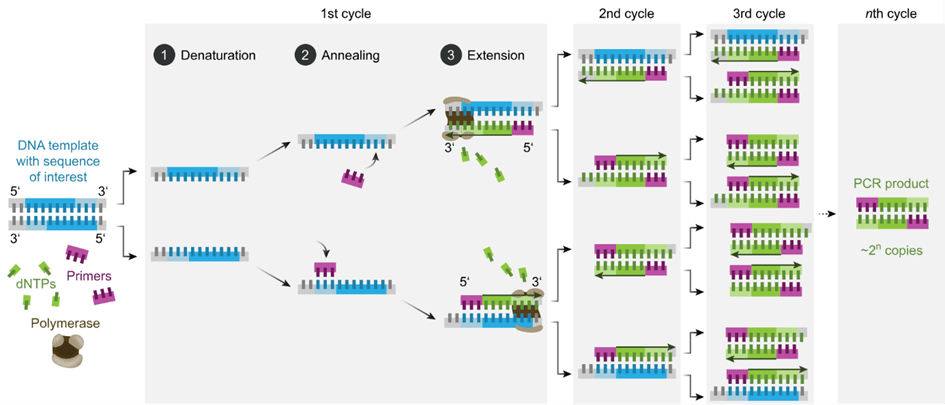
DNA amplification reaction | PCR amplification works in ~25-35 cycles of DNA denaturation (~90 °C), primer annealing (~60 °C) and DNA amplification (~72 °C). During each cycle the amount of target DNA approximately doubles (Figure 2). When designing a new PCR assay, it is usually necessary to optimise the amplification protocol, i.e. duration and temperatures of the individual steps, number of cycles and potential additives. The amplification reaction is done in a thermocycler.
Figure 2: Molecular principle of PCR (graphic from [6])
DNA product | Depending on the length of the target DNA, successfulPCR results in ~1 million copies[7] of DNA product per hour that is exactly as long as the target sequence (including the primer binding sites).
Product detection | In the basic version of PCR, the presence of the product is detected by gel electrophoresis. As this is almost never done in pathogen diagnostics these days, I won’t explain the details here. Instead, the most common detection method – quantitative PCR – is described in the next section.
Relevant PCR variations
Quantitative or real-time PCR(qPCR, unfortunately sometimes also abbreviated as RT-PCR, see "reverse transcription PCR" below) allows real-time detection of the amount of dsDNA and saves 1-2 hours compared to detection through gel electrophoresis. qPCR is done:
by adding a fluorescent dye to the reaction, which is activated by binding to dsDNA, or
by adding fluorescently labelled nucleotides, which are activated when the polymerase adds them to newly amplified dsDNA. The latter is more commonly used and more specific[8].
Measuring the fluorescence level after each PCR cycle allows to detect the amplified product and thus determine the presence or absence of target DNA in a sample. Further, the approximate viral load can be quantified via fluorescence intensity. qPCR requires a specialised thermocycler that can detect fluorescence levels of each sample.
Reverse transcription PCR (RT-PCR) is one of the most commonly used variants that allows PCR with RNA instead of DNA as a starting material. RT-PCR is especially relevant as many viruses (e.g. Corona, Influenza, Zika and Ebola viruses) contain RNA as genetic material. RT-PCR works by adding the enzyme reverse transcriptase to the reaction that copies RNA into DNA. Afterwards, PCR can be done as described above.
qPCR is today’s gold standard in pathogen diagnostics and can be combined with RT-PCR to detect RNA viruses (RT-qPCR).
Dry reagent-based PCR (DRB-PCR) allows freeze-drying the reagents in a reaction tube, which can then be kept at ambient temperature. Freeze-dried PCR reagents have been shown to be stable at 18-25 °C for up to two years[9]. To activate the reaction, water and the patient sample are added and mixed.
Nested PCR is a variant used to increase PCR specificity. A second pair of primers framing the target DNA is designed. Two subsequent PCRs are performed, the first one using the two outer primers (further away from the target), the second with the inner primers. I mention this here, because using more than two primers is one of the reasons for LAMPs high specificity (see next section). While nested PCR allows for some degree of the advantage of using two primer pairs, it slows down the pathogen detection process and is rarely used in diagnostics.
2.2 LAMP
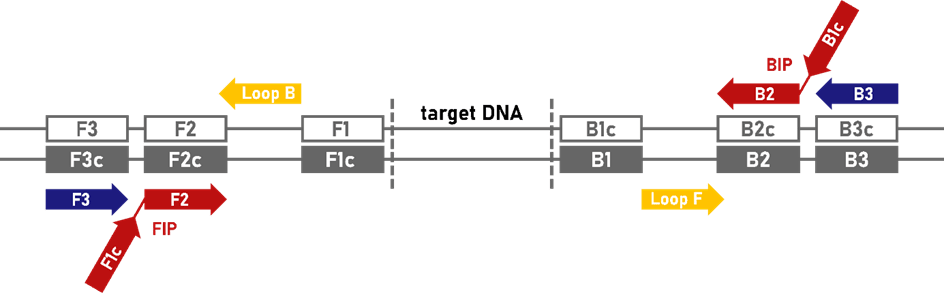
Primer design | Two to three pairs of primers are needed for LAMP (Figure 3). Framing the template, three primer-binding regions are identified. Primer design for LAMP is usually done with a primer design tool[10],[11] and is quite hard to do manually.
Inner primers (forward inner primer FIP / backward inner primer BIP, red) | These are composite primers consisting of the F2 and F1c / B2 and B1c sequences. The F1c and B1c parts of the primers are overhangs in the first reaction step, i.e. they do not bind to the DNA. The F1c and B1c sequence is thus added to the amplified DNA, which allows loop formation (self-priming) in the subsequent step.
Outer primers (F3/B3, blue) | These primers initiate DNA amplification that separates the strand synthesised from the FIP/BIP, so that the loop can form, and amplify the target itself. The outer primers additionally amplify the target without loop formation.
Loop primers (Loop F/Loop B, yellow, optional) | These are additional primers binding in the loop region (between F1/F2 and B1/B2 regions). Their use can more than halve the reaction time of LAMP[12] as the number of starting points for the polymerase is increased.
Figure 3: Primers for LAMP. Red = inner primers, blue = outer primers, yellow = loop primers (optional), grey = binding regions on DNA, F = forward, b = backward, c = complimentary (graphic adapted from [13])
DNA extraction is not necessarily needed, the LAMP polymerase is much more robust towards inhibitors present in unpurified samples than the ones used in PCR[7].
Reagents | The reason why LAMP can be done isothermally (i.e. at one constant temperature) is that the DNA polymerase used for LAMP has a strand displacement activity: It can separate a DNA double-strand while synthesising a new complementary strand (auto-cycling) which eliminates the need for a separate denaturation step. As DNA denaturation starts at 60-65 °C, allowing parts of the dsDNA to separate, the primer binding can still happen, and any residual dsDNA is displaced by the LAMP polymerase.
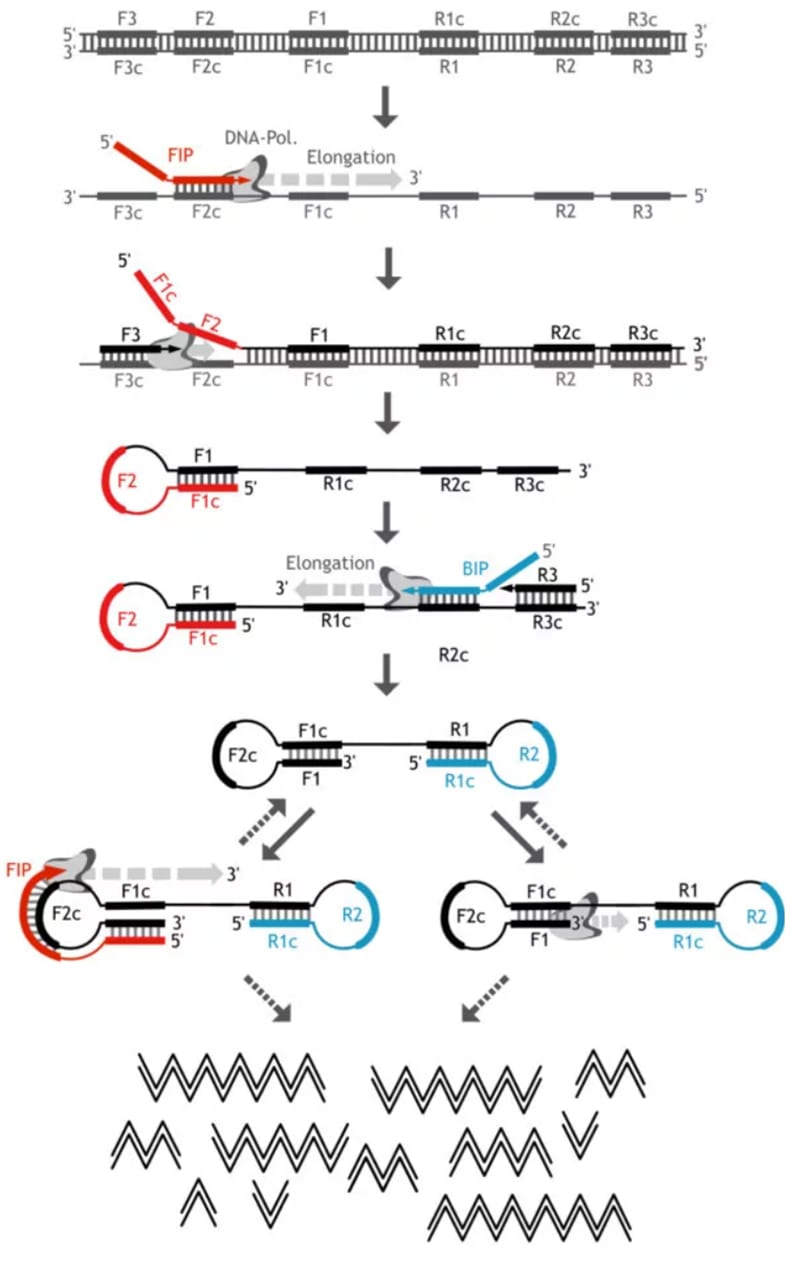
DNA amplification reaction | Amplification is usually done at constant ~60-65 °C for ~20-40 minutes. The required equipment is a simple heated water bath, incubator or dry block heater. Even the use of a thermos bottle[14] or standard household oven[15] as incubators have been described. The reaction mechanism following the first amplification steps (outlined in the “primer design” section above) are shown in Figure 4. For a nice explanation of the individual steps, see this video:
Figure 4: Molecular Principle of LAMP (graphic adapted from [13])
DNA product | Successful LAMP results in way over a billioncopies of DNA product per hour. The product contains multiple copies of the target sequence in chains of different length separated by the primer binding sites. Due to the loop-formation on the primer binding sites, these DNA chains form “cauliflower-like” structures[4].
Product detection: analysis with naked eye
Turbidity: Magnesium pyrophosphate (MPP) precipitates as a side product of massive DNA amplification and turns the solution opaque. Turbidity can be analysed one with the naked eye or with a photometric detection device (that is simpler than the one needed for fluorescence detection like in qPCR). A combination with real-time analysis is possible. (Turbidity analysis does not work in PCR, because MPP concentrations don’t become high enough to create visible precipitation and MPP hydrolyses at 95 °C[7].)
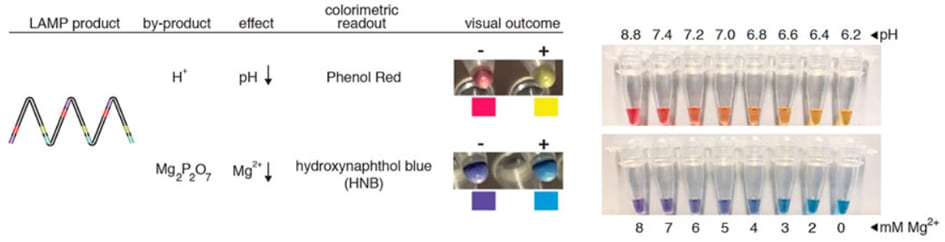
Colour change detection includes:
pH-dependent dye indicators (e.g. phenol red), that induce a colour change as the pH value of the reaction decreases upon DNA amplification. This is one of the most used dyes in LAMP assays[15],
metal ion indicators such as hydroxynaphthol blue, that change colour with dropping free magnesium ions as MPP accumulates through DNA amplification[15].
Figure 5: Colorimetric detection of LAMP products via phenol red and hydroxynaphthol blue (graphic from [15]).
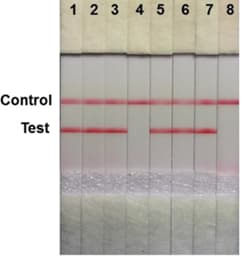
Lateral flow (dip-)stick (LFD-LAMP) detectionis similar to the SARS-CoV-2 antigen tests many of us have used during the pandemic. A lateral flow stick is dipped into the product (or LAMP product is applied directly to the stick) and the result can be read out in a few minutes: a negative test shows one coloured line, a positive test two lines. This detection method requires a relatively inexpensive modification of two primers[14] to facilitate binding to the control and test lines on the flow stick.
Figure 6: Schematic of eight lateral flow (dip-)sticks, samples 4 & 8 show a negative result, the remaining samples a positive one (graphic from [14])
Product detection: further equipment needed
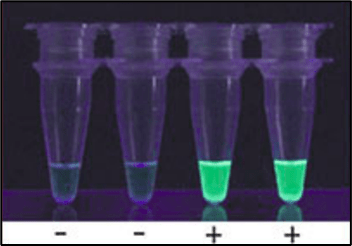
Fluorescence-based detection requires a device that simultaneously allows fluorescence read-out. In simple cases, this can be a UV-lamp in a dark space that allows fairly easy read-out with the naked eye. More sophisticated (and costly) appliances combine the heated block with a real-time fluorescent read-out[16]. The most common fluorescence-based detection methods for LAMP are:
dsDNA-intercalating dyes (like SYBR-Green), that bind preferably to dsDNA,
calcein, which is a fluorescent dye that is added to the reaction in its inactive form, loaded with manganese. Upon amplification, the manganese starts forming complexes with MPP, releases calcein and thereby activates fluorescence.
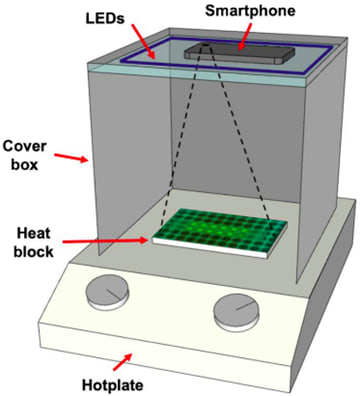
smaRT-LAMP[17] (Figure 8) is a smartphone-based calcein detection method that uses the camera and external LEDs in a dark box to detect fluorescence.
Figure 7: Visual detection of SYBR-green fluorescence in UV-light (graphic adapted from [18])
Quantitative or real-time LAMP, reverse transcription LAMP (RT-LAMP) and dry reagent-based LAMP (DRB-LAMP) are in their principle analogous to the respective modifications in PCR.
2.3 Summary of key differences between PCR & LAMP
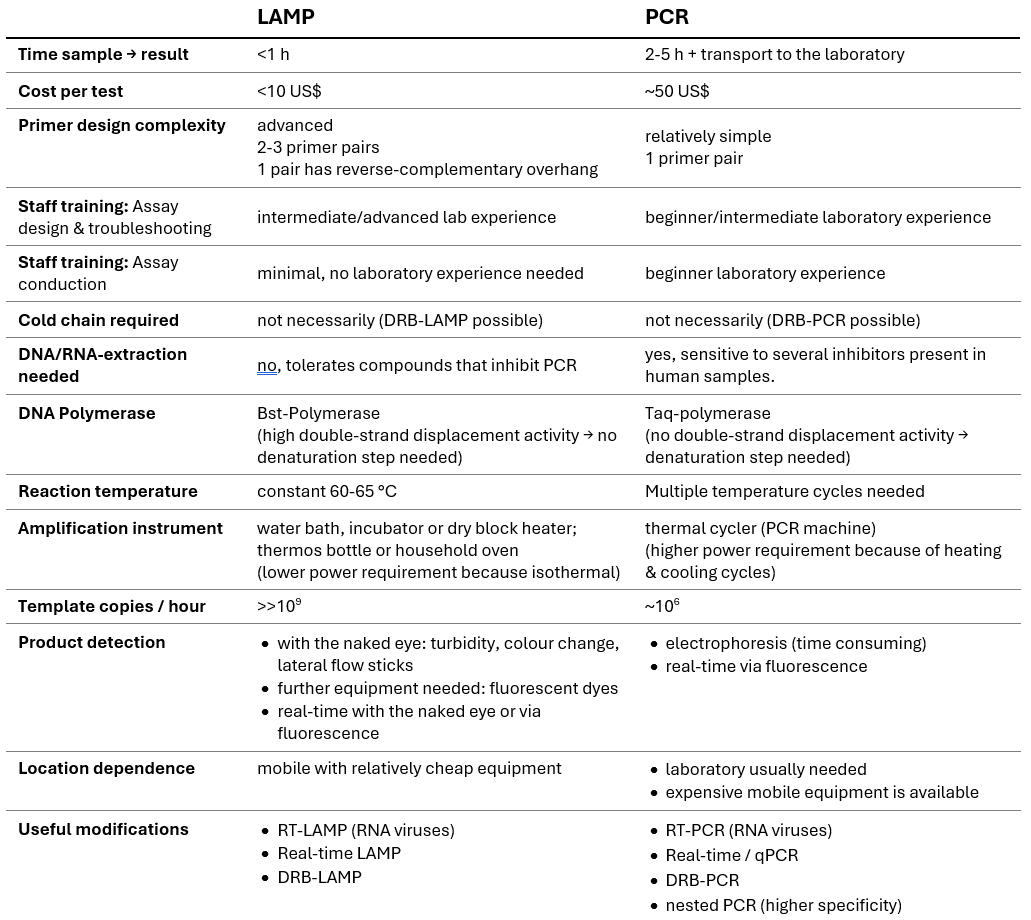
Table 1: Comparison of LAMP and PCR used for infectious disease detection. (DRB = dry reagent based, RT = reverse transcription, qPCT = quantitative polymerase chain reaction)
LAMP has several advantages over PCR, especially in resource-constrained settings. It is 2-5x faster, ~5x cheaper and far more robust. The low-tech equipment and simple product detection methods facilitate point-of-care diagnostics and assay conduction by minimally trained staff.
Still, we neither widely adopted LAMP tests during the SARS-CoV-2 pandemic nor is LAMP predominantly used in biomedical research. Here are the reasons I identified for why this is the case.
3. Why is LAMP not used more widely?
3.1 Complexity of LAMP mechanism, assay design & optimisation
The molecular mechanism of LAMP is more complex and thus harder to understand than the one of PCR[19]. This is due to:
the use of more primers (4-6 vs. 2), two of them being composite overhang primers (FIP/BIP),
the mechanism of loop formation through the composite overhang primers that introduce a sequence that is reverse complementary to an adjacent region on the template DNA, thus forming the loops after elongation from the first overhang primer,
amplification starting at 4-6 different primers,
simultaneous amplification from many starting points, which is not separated through cycles due to the strand displacement activity of the DNA polymerase,
multitude and variety of DNA products due to interaction of different primers and chaining of the target DNA.
Generally, one could say LAMP is “messier” than PCR.
The complexity of the method comes with bigger challenges for the initial design of a LAMP assay for a certain target DNA. Compared to PCR, the primer design is more difficult and harder to do manually4,18. The presence of more primers increases the risk of primers binding to each other (forming “primer dimers”) instead of the target DNA. Longer primers, such as the inversion primers are more prone to self-binding and forming a “hairpin”. Primer dimers and hairpin structures can provide double-stranded starting points for the DNA polymerase, which can lead to unspecific DNA amplification and thus false-positive test results[20].
In summary, understanding the mechanism and designing and optimising a new assay are harder for LAMP than for PCR and much harder than conducting a pre-designed LAMP assay (i.e. testing someone for a pathogen of interest).
3.2 Lower versatility of LAMP
The invention of PCR in the early 1990s allowed something that wasn’t possible before - DNA amplification – and facilitated a plethora of subsequent research. Seven years later, LAMP offered another method for DNA amplification, that certainly improved relevant aspects of PCR, but is also much less versatile than PCR, so the degree of innovation was lower.
LAMP excels in diagnostic applications, where the “research” question – whether the tested individuum is infected with a certain pathogen or not – is answered through the presence or absence of an amplification product. Besides diagnostics, PCR often (probably predominantly) functions as an initial and/or intermediate step in longer research questions. PCR products are often used for further analyses such as cloning or sequencing. While it is not generally impossible to use LAMP product for these applications, the general structure of LAMP amplicons (various lengths and chained target DNA) is much less suitable than the clean PCR amplicons that are of one defined length and contain one copy of the target DNA each.
I assume the lower degree of innovation and the lower versatility - and thus fewer use cases - of LAMP are a key reason why it is generally less well known and thus also less widely used for diagnostics, where it could theoretically outperform PCR.
3.3 PCR is a well-established standard
A search on PubMed (a meta database for research literature on life sciences and biomedical topics) yields about 650k publication results for ‘polymerase chain reaction’ (200k in the last ten years) and only 5k results for ‘loop-mediated isothermal amplification’ (4k in the last ten years).
PCR is profoundly well-researched and validated which makes it the default choice in many laboratories. In (clinical) diagnostics, where false positives or negatives can have serious consequences, there are often standards to adhere to, which further limit adoption of novel protocols. Most laboratories are equipped with thermocyclers, staff is trained for PCR methods, the supply chain for reagents is well established and many pre-designed assay kits that have undergone standardisation and quality control, are available.
While many publications have outlined LAMPs potential with comparable and sometimes better accuracy than PCR, there are also numerous reports with lower accuracy [21]. It seems to me that there is significant room for improvement through further research, and I would assume that with further research, LAMP can become a much more powerful and widely used diagnostic tool. I consider this particularly important as the firm establishment of PCR in diagnostics (and biomedical research at all) is most prominent in the industrialised world.
3.4 Lower commercial incentives for offering LAMP assays
Why hasn’t the market solved it[22]? As PCR is already highly established in the industrialised world, introducing a new method (against general conservatism and opposition to change) offers less financial incentives for companies, especially if the new method is much cheaper. Similarly, low- and middle-income countries might not pose a lucrative enough market. This would create the need for either state actors or philanthropy to step up.
3.5 Public funding gap for pandemic preparedness, inertia in state actors and a need for public policy advocacy
Why hasn’t the state solved it[22]? Compared to the human lives and wellbeing, economies, knowledge etc. that are at stake, global investment of public resources into pandemic preparedness and response is sparse, which is part of why this cause area is high on the agenda of 80,000 hours’ world’s most pressing problems2.
The SARS-CoV-2 pandemic raised awareness for the importance of and started concrete public actions towards pandemic preparedness and response. The pandemic also gave rise to several efforts at improving LAMP assays and spawned a few promising candidates[21]. Yet these advancements possibly came to light too far into the pandemic when testing systems were already well-established and any disruptions may have seemed more of a hassle than a benefit[21].
Centralised funding, approval and promotion of fast, cheap, robust, general and scalable tests is important for preventing and containing infections. It remains neglected and creates a need for public policy advocacy.
3.6 LAMP - A potential focus area for philanthropy?
Why hasn’t philanthropy solved it[22]? While there are a number of possible interventions to promote - further research, identifying commercial incentives, and public advocacy – I could also imagine some of LAMPs untapped potential addressed in non-profit settings. I haven’t found any previous efforts in this direction. Here are the areas I’d consider relevant to work on:
identifying the needs for different tests and specific test properties,
advocating for pandemic preparedness and cooperating with governments and disease control agencies,
and finally designing assays and making them available or facilitate local production.
Soroka, M., Wasowicz, B. & Rymaszewska, A. Loop-Mediated Isothermal Amplification (LAMP): The Better Sibling of PCR? Cells 2021, Vol. 10, Page 193110, 1931 (2021).
Kralik, P. & Ricchi, M. A basic guide to real time PCR in microbial diagnostics: Definitions, parameters, and everything. Front Microbiol8, 239909 (2017).
Nagamine, K., Hase, T. & Notomi, T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol Cell Probes16, 223–229 (2002).
Yang, Z., Xu, G., Reboud, J., Kasprzyk-Hordern, B. & Cooper, J. M. Monitoring Genetic Population Biomarkers for Wastewater-Based Epidemiology. Anal Chem89, 9941–9945 (2017).
Kellner, M. J. et al. A Rapid, Highly Sensitive and Open-Access SARS-CoV-2 Detection Assay for Laboratory and Home Testing. Front Mol Biosci9, 801309 (2022).
Heithoff, D. M. et al. Assessment of a Smartphone-Based Loop-Mediated Isothermal Amplification Assay for Detection of SARS-CoV-2 and Influenza Viruses. JAMA Netw Open5, e2145669–e2145669 (2022).
I have >5 years of lab experience working with PCR and its variations. I have designed and optimised multiple PCRs and I did have a bit of a hard time understanding LAMP at first.
Meagher, R. J., Priye, A., Light, Y. K., Huang, C. & Wang, E. Impact of Primer Dimers and Self-Amplifying Hairpins on Reverse Transcription Loop-Mediated Isothermal Amplification Detection of Viral RNA. Analyst143, 1924 (2018).
BLUF:
* To determine whether AI is ‘improving exponentially’, ‘hitting the wall’, or any other claim which involves a quantity or magnitude (e.g. ‘This model was a big leap/small increment’). We need a good y-axis: an interval scale of AI capability which means +1 unit always represents the same degree of ‘how much better’, in the same way +1 degree Celsius is always the same amount of ‘how much hotter’.
* Yet there is no good y-axis for AI capability. All our...
Summary
* The animal welfare movement has already seen an influx in funding and should prepare for the possibility of more.
* The EA Animal Welfare Fund is encouraging those working in animal advocacy to actively set aside time and resources now to concretely plan for scaling sustainably, and we’ll support you in doing that.
* We’re requesting advocates set concrete ambitious goals and submit plans t...
Public service announcement
1. Applications are now open for our first ever round of the Charity Entrepreneurship Incubation Program dedicated exclusively to animal welfare. Learn more about what’s different this round here and apply...



{kind=link}