Cross-posted from my NAO Notebook.
This is an edited transcript of a talk I just gave at CBD S&T, a chem-bio defence conference. I needed to submit the slides several months in advance, so I tried out a new-to-me approach where the slides are visual support only and I finalized the text of the talk later on. This does mean there are places where it would be great to have additional slides to illustrate some concepts
Additionally, this was the first time I gave a talk where I wrote out everything I wanted to say in advance. I think this made for a much less engaging talk, and for future ones I'm planning to go back to speaking from bullets.

I'm Jeff Kaufman, and I co-lead the Nucleic Acid Observatory at SecureBio, where we work to develop biosurveillance methods capable of detecting biological threats released by governments or non-state actors. Today I'm going to be talking about the computational methods we use in our pathogen-agnostic early warning system.
Before I get started, however, this is all the work of a team:

I'm honored to work with this group, and Will, Lenni, and Simon are here today.
As I go through this talk, please leave questions in the app. If I've managed my timing correctly, always a risk, we should have a good bit of time for them at the end.
So: what are we up against?
A typical pandemic starts with an infected individual.

They start having symptoms and become infectious.

Someone else gets infected.

Then a few more.

Those people start having symptoms and become infectious.

More people get infected.

There is now a cluster of unusual cases.

Perhaps at this point doctors notice something unusual is happening and raise the alarm.
But what if there's a long pre-symptomatic period? Imagine something like a faster-spreading HIV, maybe airborne.

You'd still start with one infected individual.

Then another.
Still without the symptoms that would tell us something was wrong.

No one notices.

Then the first person starts showing symptoms

But that's unlikely to be enough for someone to notice something is wrong: there are individuals who report unusual symptoms all the time.
Another person starts showing the same unsual symptoms.

But they're far enough away that no one puts the pieces together yet.
By the time you have symptom clusters large enough to be noticed...

A very large fraction of the population has been infected with something serious.
We call this scenario a "stealth" pandemic.
I've talked about this as presymptomatic spread, but note you'd also get the same pattern if the initial symptoms were very generic and it just looked like a cold going around.
Now, barriers to engineering and synthesis are falling.
There are dozens of companies offering synthesis, many with inadequate screening protocols. And benchtop synthesizers are becoming more practical.
Transfection, to get synthesized DNA or RNA into an organism, is also getting more accessible as more students learn how to do it and the knowledge spreads.
And frontier AI systems can provide surprisingly useful advice, helping an attacker bypass pitfalls.
What if a stealth pandemic were spreading now?

Could our military personnel and general public already be infected? How would we know?
We probably wouldn't. We are not prepared for this threat. How can we become prepared?
We need an early warning system for stealth pandemics. We're building one.

We're taking a layered approach, starting withtwo sample types: wastewater
and pooled nasal swabs.
We extract the nucleic acids, physically enrich for viruses, and run deep untargeted metagenomic sequencing.
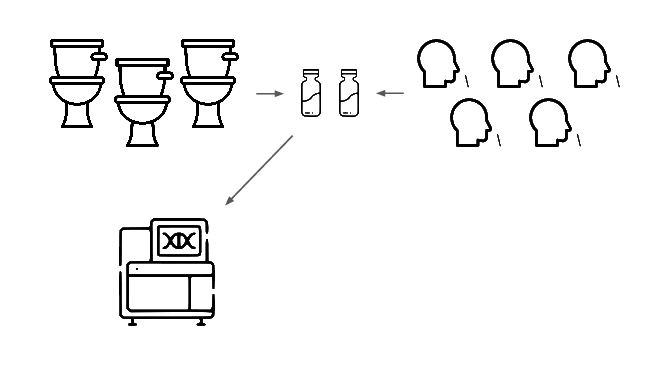
Then we analyze that data, looking for evidence of genetic engineering.
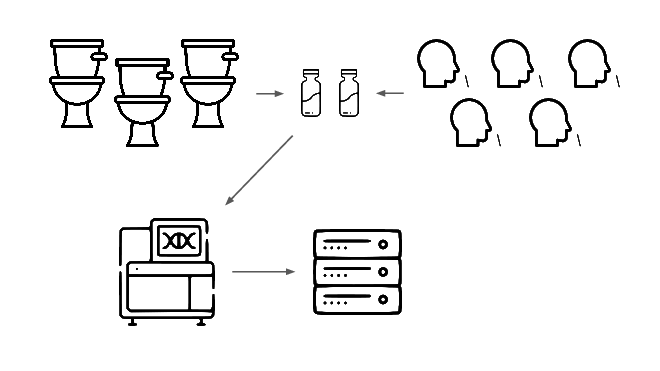
When this flags a cluster of sequencing reads indicative of a potential threat, analysts evauate the decision and the data the decision was based on.
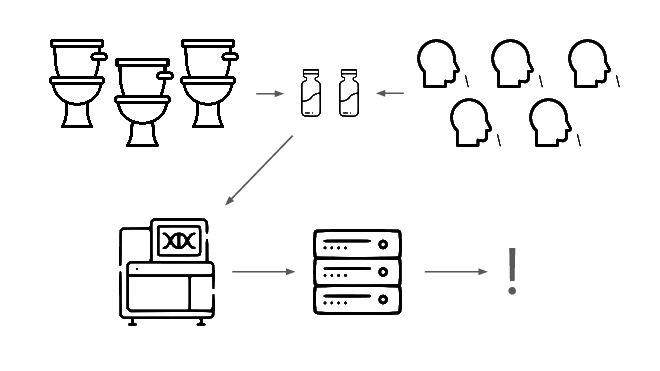
Lets go over each of these steps in more detail.
Where and what should we sample?

We've done the most work with municipal wastewater. It has some serious advantages, but also serious disadvantages.
The best thing about it is the very low cost per covered individual.

A whole city, in a single sample.
On the other hand, only a tiny fraction

of the nucleic acids in wastewater come from human-infecting pathogens, and even fewer come from non-gastrointestinal viruses.
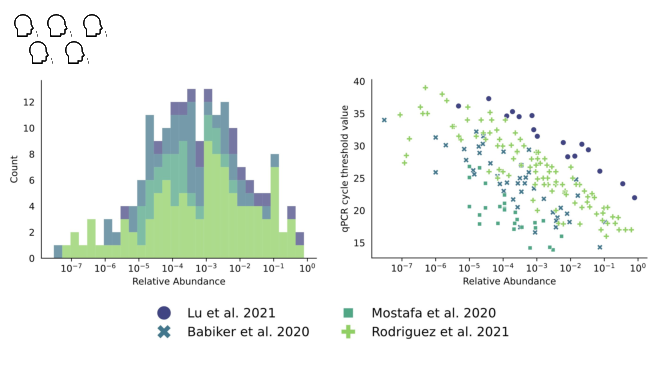
We looked into this in a paper I link at the end. We were trying to estimate the
fraction of sequencing reads that might come from covid when 1% of people had been infected in the last week.
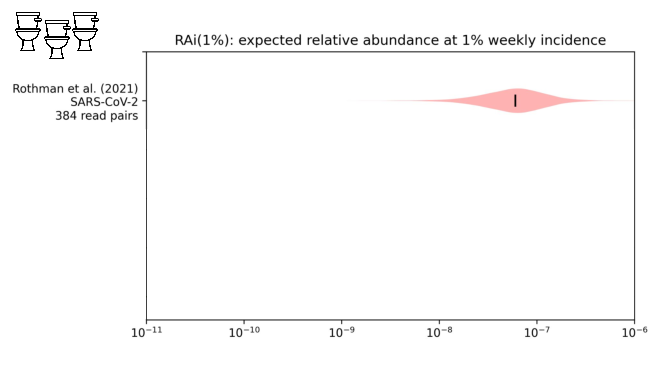
While our distribution is still wide, we estimate about one in ten million sequencing reads would come from covid.
With influenza, however...
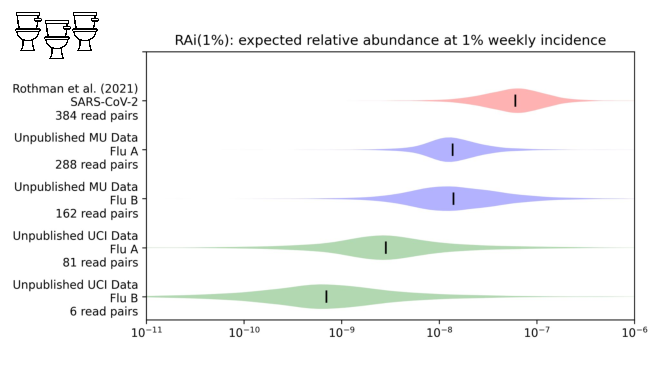
It's one to two orders of magnitude lower.
You need a lot of sequencing to see something that's only one in 100M reads!
On the other hand, bulk sequencing keeps getting cheaper.
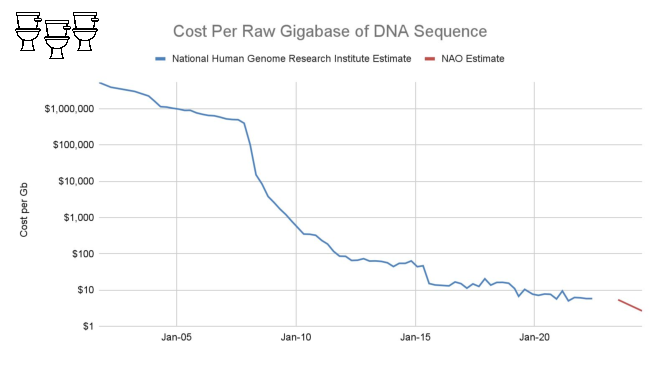
The list price of a NovaSeq 25B flow cell is $16k for about 18B read pairs, or about $900 per billion. This is low enough to make an untargeted sequencing approach practical.
The largest cost at scale, however, is still sequencing.
You still need a lot of sequencing.
Very deep sequencing.
We are sequencing a typical sample to between one and two billion reads.
For an approach with the opposite tradeoffs we're also exploring pooled nasals swabs.

We do anterior nasal, like a covid test.
With swabs, the fraction of sequencing reads that are useful is far higher.

We looked into this in our swab sampling report, which I'll also link at the end.
With covid, a swab from a single sick person might have one in a thousand sequencing reads match the virus.
Instead of sampling a single person, though...

We sample pools. This is a lot cheaper, since you only need process a single combined sample.

We estimate that with a pool of 200 people
, if 1% of the population got covid in the last week approximately one in 5,000 sequencing reads would come from the virus.
Because relative abundance is higher, cost-per-read isn't a driving factor. Instead of sending libraries out to run a big Illumina sequencer...
We can sequence them on a Nanopore, right on the bench.

The big downside of pooled nasal swab sampling, though, is you need to get swabs from a lot of people.

A lot of people.

A lot a lot.

We go out to busy public places and ask people for samples.

We get about 30 swabs per hour and are running batches of 25-30 swabs. At this scale this isn't yet cost-competitive with wastewater sequencing as a way to see non-GI viruses, but as we keep iterating on our sample collection and processing we're optimistic that we'll get there.
So you collect your samples and sequence them to learn what nucleic acids they contain. What do you do with that data?
How do you figure out what pathogens are present?

The traditional way is to match sequencing reads against known things.

It's great that sequencing lets us check for all known viruses that infect humans, or that could evolve or be modified to infect humans. But this isn't pathogen agnostic because it doesn't detect novel pathogens.
Our primary production approach is a kind of genetic engineering detection.
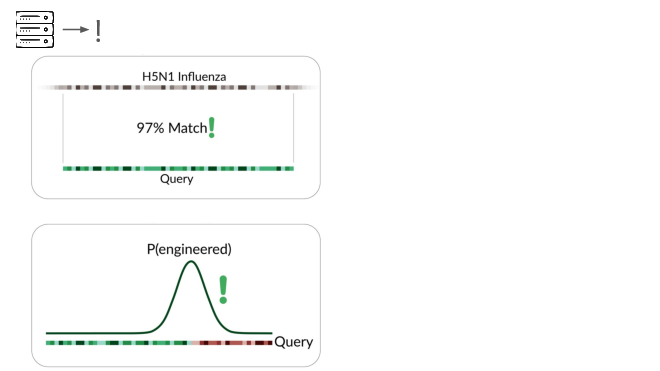
If someone were trying to cause harm today, they'd very likely start with something known. Many ways of modifying a pathogen create a detectable signature:

A junction between modified and unmodified portions of a genome. We can look for reads that cover these junctions.
Here's a real sequencing read we saw on February 27th
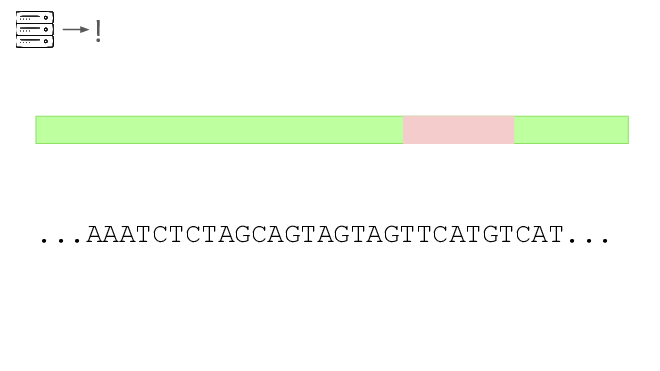
The first part is a solid match for HIV
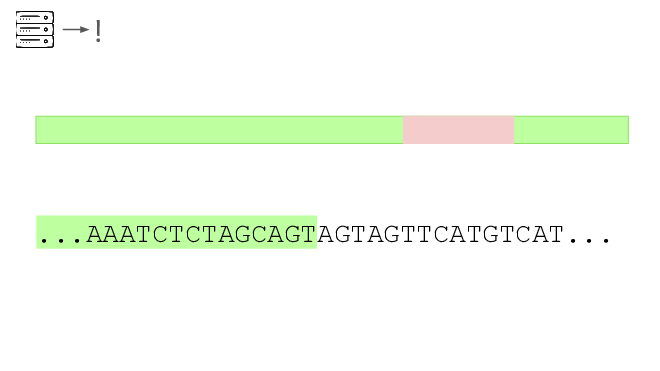
But the second part is not.
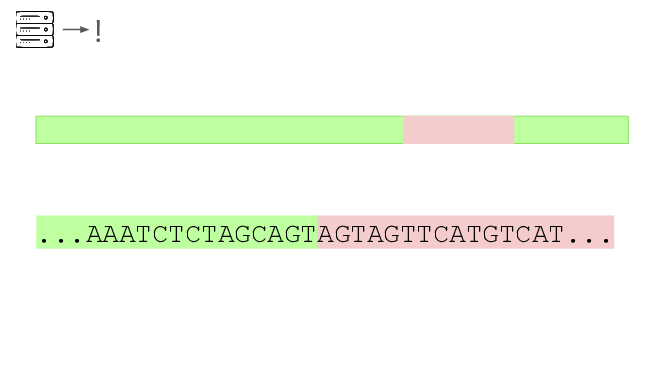
This is a common lentiviral vector, likely lab contamination. But it's genetically engineered, and it's the kind of thing the system should be flagging. It has detected three of these so far, all derived from HIV.
To validate this approach of looking for surprising junctions we started in silico

We picked a range of viruses, simulated engineered insertions, simulated reads from these genomes and verified the system flags reads. This came in at 71% of optimal performance.
Then we validated real world performance with spike-ins. We took raw municipal sewage...
And added viral particles with engineered genomes.
With three different HIV-derived genomes we flagged 79-89% of optimal performance.
In addition to detecting pathogens via...
direct match to known things and by flagging suspicious junctions, we can flag based on growth patterns.
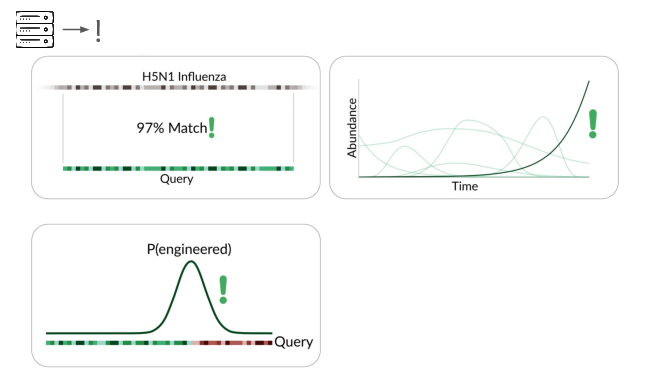
Initially we're taking a reference-based approach, detecting known things becoming more common. This can be more sensitive than junction detection because you don't have to see the junction to know something's wrong. This means a larger fraction of sequencing reads are informative.
Because this method is based on known things becoming more common, it's far more sensitive in cases where the base genome is initially rare or absent in your sample type. But this combines with other detection methods to further constrain the adversary's options.
We've also explored a reference-free approach, looking for exponentially-growing k-mers. This requires more data than we have so far, but it's the hardest one for an adversary to bypass: it tracks the fundamental signal of a pathogen spreading through the population.
The final method we're exploring is novelty detection.
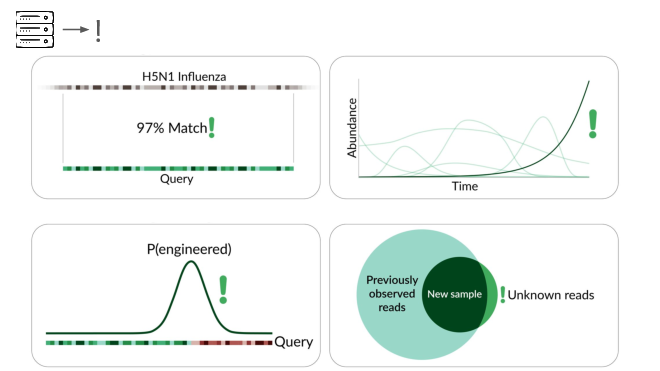
If we can understand these samples well enough, we can flag something introduced just for being unexpected in the sample. This is a major research problem, and we're excited to share our data and collaborate with others here. For example, Willie Neiswanger and Oliver Liu at USC have trained a metagenomic foundation model on this data, which they're hoping to apply to detecting engineered sequences. We're also collaborating with Ryan Teo in Nicole Wheeler's lab at the University of Birmingham on better forms of metagenomic assembly.

How big would a system like this need to be? It depends heavily on our desired sensitivity.

To detect when 1:100 people are infected

We need a medium amount of wastewater sequencing
Or swab collection.
To detect

when 1:1,000 people have been infected

We'd need 10x the sequencing

Or 10x the swabbing

We've made a simulator for estimating the sensitivity of different system designs.
Given a system configuration, it estimates how likely it is to flag a novel pathogen before some fraction of the population has ever been infected
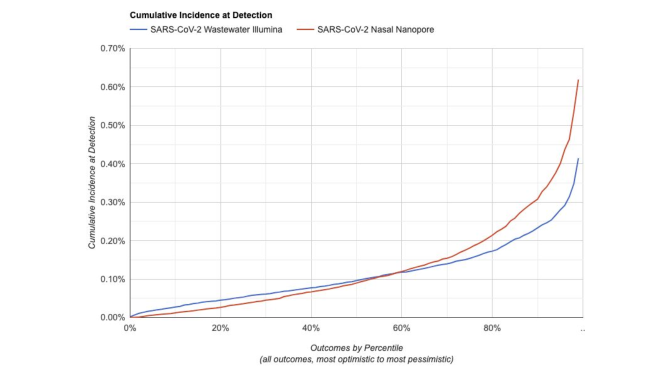
For something in the range of $10M/y we think you can operate a system capable of detecting a novel pathogen before 1:1000 people have been infected. But there is still a lot of uncertainty here, and we'll learn more as we continue our pilot.

Today I've described an approach to pathogen-agnostic detection, ...
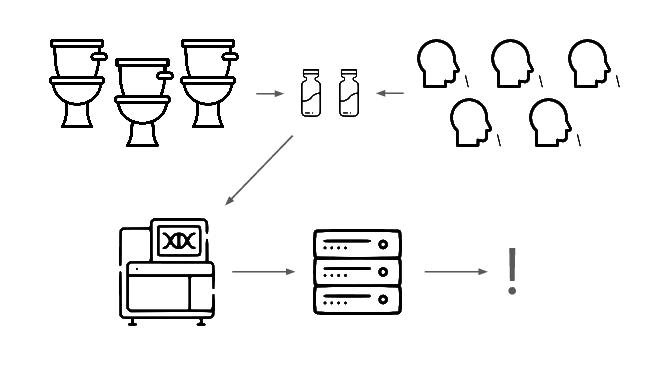
Starting with wastewater and nasal swabs, extracting and sequencing the nucleic acids, and then applying a range of computational methods to identify engineered pathogens.
This is a system that we need to defend against stealth pathogens, and we're building it. Some parts are ready to go today, such as junction-based genetic engineering detection. Other parts we're close to, and are a matter of engineering and tuning, such as reference-based growth detection. And others need serious research, such as understanding complex samples so well that we can flag things for being new. We're excited to be working on this, and we're interested in collaborating with others who are thinking along similar lines!
Thank you.
I've prepared a webpage with links to the resources I discussed in this talk:

We have some time for live questions

And I'm also happy to take questions over email at jeff@securebio.org

Comment via: facebook, lesswrong, the EA Forum, mastodon, bluesky
I found this a really clear and useful explanation (though I already had a decent idea how NAO worked)!
If ever you want to reach a broader audience, I think making an animated video based on this content, maybe with the help of Rational Animations or Kurtzgesagt, would work well.
Assuming a key inefficiency of the nasal swabs method is the labour costs of people collecting them, is the process straightforward enough that you could just set up an unmanned sample collection place where in a busy building somewhere people can just swab themselves and drop the sample in a chute or box or something? Hopefully post-Covid people are fairly familiar with nasal swabbing technique.
I'm finishing up a video covering NAO and wastewater monitoring, though not based on this specific talk.
Wow!
If you'd like me to review it for accuracy before you publish it I'd be happy to!
I think the main downside of setting up a sample collection box is that the samples would probably sit a lot longer before being processed, and RNA degrades quickly. I also suspect you wouldn't get very many samples.
(The process itself is super simple for participants: you just swab your nose.)
This was a very informative! I was familiar with NAO, but I had no idea how low the RNA concentration is of respiratory viruses in wastewater, and how much that drives up the cost of sequencing. How do indoor aerosol samples compare? Seems like air sampling of indoor public spaces might be promising, I found a couple papers demonstrating that detecting pathogens via airborne MGS sequencing is possible but none gave relative abundance numbers.
Executive summary: SecureBio's Nucleic Acid Observatory is developing an early warning system to detect engineered pathogens before they cause widespread infection, using a combination of wastewater sampling, pooled nasal swabs, and advanced computational analysis methods.
Key points:
This comment was auto-generated by the EA Forum Team. Feel free to point out issues with this summary by replying to the comment, and contact us if you have feedback.